Introduction
Adding enzymatic parameters to a metabolic network model helps to improve the accuracy of simulations, to understand underlying mechanisms of reactions and to predict dynamic responses of the system to environmental changes. These enriched models facilitate optimization of experimental conditions, validation and calibration of predictions, and the identification of potential therapeutic targets. They can also be used to analyze the effects of perturbations, such as mutations or drug inhibitors, on metabolic fluxes.
Although there are several very important parameters such as Michaelis-Menten constants (Km), optimum pH etc., we have chosen to focus on the catalytic constants (turn-over) also known as kcat. Indeed, kcat are directly involved in metabolic fluxes, by quantifying the maximum speed at which an enzyme catalyzes a reaction, thus providing an essential measure for modeling and understanding the dynamic behavior of metabolic networks.
In current models, the reaction flux limits are not always biologically realistic, as they are often based on standard or approximate values. This can lead to inaccurate predictions of metabolic fluxes and network behavior. By using kcat values, it is possible to better bound these fluxes and provide more relevant results. kcat constants can be used to define more precise flux limits (lower and upper bounds), better reflecting actual enzyme capacities under specific conditions.
These enzymatic data can be found directly in Gencovery’s BIOTA database. BIOTA is a unified and structured collection of omics data collected from official open European (EMBL-EBI) and NCBI taxonomy knowledge bases. More than 2 M organisms are referenced in BIOTA with their metabolic characteristics.
Prerequesites
- An acces to Constellab and to a digital lab
- The brick “gws_biota” (version >= 0.7.2)
- The brick “gws_gena” (version >= 0.8.0)
Steps to follow
- Create a new experiment
- Import “Load BiGG Models” task to download the metabolic network model of your choice (using the name or model ID currently on BiGG Models)
- Import “Retrieve Kcat from BIOTA” task, which retrieves all the ec numbers for the species selected and associates them with the median kcat value. This reduces extreme variations and provides a more reliable, generic value for modeling metabolic fluxes.
- Import “EC number to Bigg ID” task which filters reactions: only reactions with an identifier on BiGG Models are retained.
- Import “Create a contextualization file (kcat)” task which builds the final file that will be used to contextualise the digital twin.
Be sure to use the same species throughout the experiment!
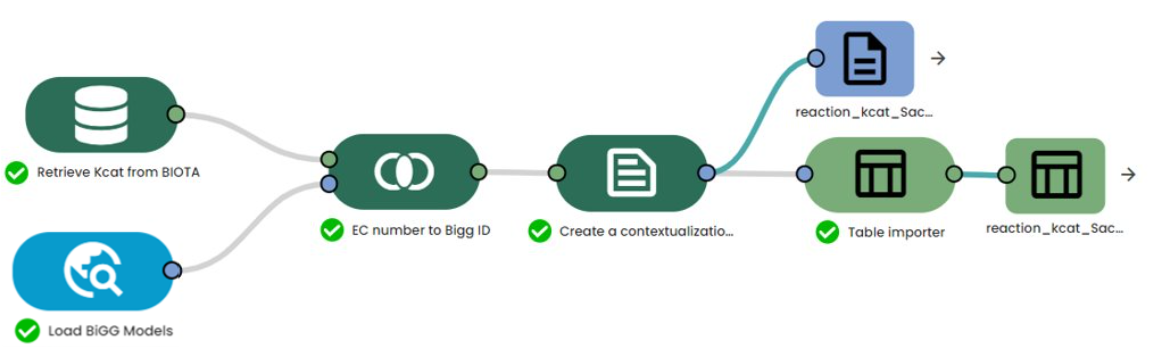
Results
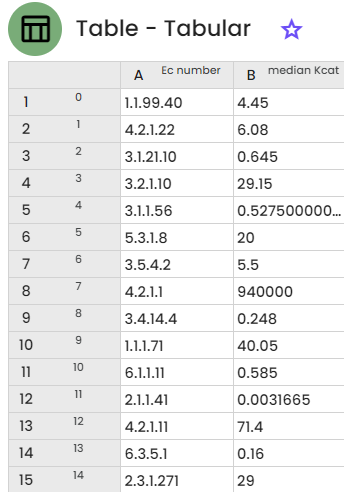
In BIOTA, we have retrieved 213 Ec numbers with available kcat values, for the species "Saccharomyces cerevisiae" (iMM904 model on BiGG Models). The first column corresponds to the ec number and the second column to the median kcat value for each ec number.
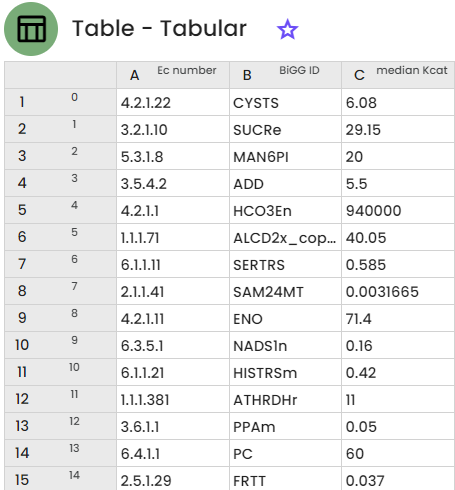
Of these 213 results, only 124 reactions have a BiGG Models identifier. Each line corresponds to an ec number with its BiGG identifier and median kcat value.
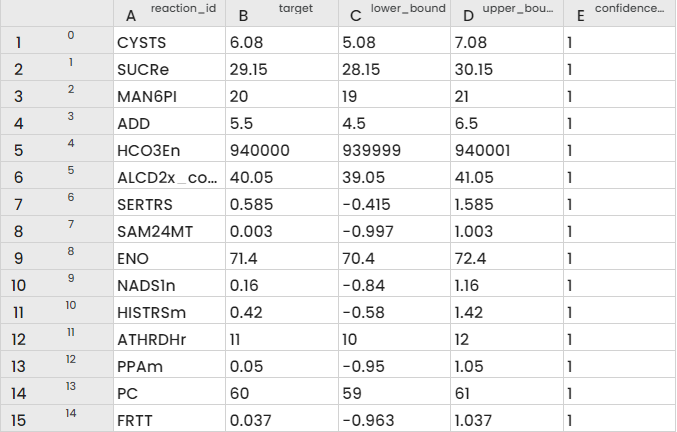
Finally, the final file can be constructed as :
reaction_id,target,lower_bound,upper_bound,confidence_score
CYSTS,6.080,5.080,7.080,1
... where “target” is the median value of kcat, “lower_bound” is calculated according to the median value of kcat - 1, “upper_bound” is calculated according to the median value +1.
To go further
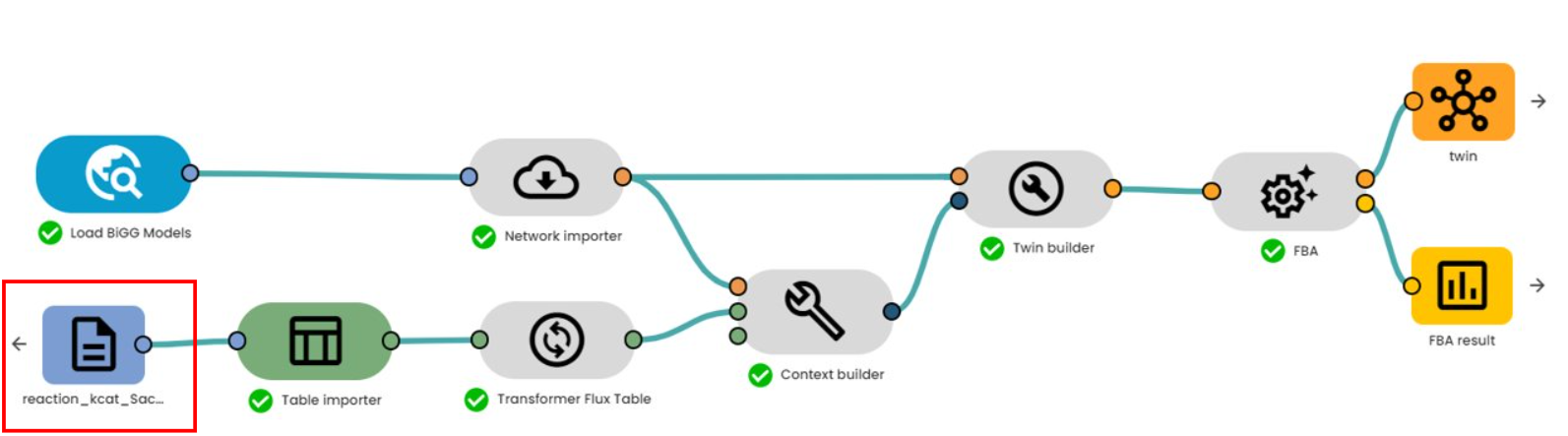
The final contextualization file created (boxed in red) can be used by a digital twin to accurately reconstruct the metabolic network (see "How to simulate digital twins of cellular metabolism from GEM models?").
This file contains the BiGG identifiers of the reactions as well as the enzymatic parameters enabling the digital twin to faithfully simulate the biological processes. Thanks to this reconstruction, subsequent analyses, such as Flux Balance Analysis (FBA), can be carried out to explore and optimize the behavior of the metabolic network in various scenarios.
Conclusion
In conclusion, the integration of enzymatic parameters, particularly kcat catalytic constants, into metabolic network models considerably improves the accuracy and relevance of simulations. This leads to a better understanding of metabolic mechanisms, optimization of experimental conditions and more reliable identification of therapeutic targets. The final contextualization file, enriched with median kcat values, can be used by a digital twin to faithfully reconstruct the metabolic network and perform in-depth analyses such as flux balance analysis (FBA), giving access to even more robust and accurate research and applications.

Comments (0)
Write a comment