Q: Is constellab pipeline adapted to ligation sequecing ?
The Constellab pipeline is not currently optimized for ligation-based sequencing workflows.
It's 16s rRNA sequencing pipeline targeting the V3–V4 regions for bacterial identification.
It's adapted for reads generated by NovaSeq, iSeq, or MiSeq sequencers.
Q: What is the difference between metagenomics and amplicon sequencing?
Metagenomic sequencing, on the other hand, involves sequencing the entire genetic material (DNA or RNA) present in a sample, without targeting specific regions. It provides a comprehensive snapshot of the entire microbial community, including both the known and unknown microorganisms. Metagenomic sequencing can identify not only the taxonomic composition of the community but also the functional potential of the microorganisms present. By analyzing the entire genetic material, researchers can study the entire microbial genome and identify the presence of specific genes and pathways involved in various functions such as metabolism, antibiotic resistance, and virulence. Metagenomic sequencing provides a higher resolution in terms of taxonomic identification and functional potential compared to amplicon sequencing but can be more computationally and financially demanding.
In summary, amplicon sequencing provides information about the taxonomic composition and relative abundance of specific microbial taxa within a community, while metagenomic sequencing provides a more comprehensive view of the entire microbial community, including taxonomic information as well as functional potential. The choice between amplicon sequencing and metagenomic sequencing depends on the research objectives, available resources, and the level of detail required for the analysis (source).
Q: What are the types of bacterial 16S rRNA ?
There are three types of bacterial ribosomal RNA (rRNA): 5S rRNA (120 bp), 16S rRNA (about 1540 bp) and 23S rRNA (about 2900 bp). 16S rRNA is commonly found in prokaryotic cells and has a high content and copy number (accounting for more than 80% of total bacterial RNA), easy access to templates, high functional homology and moderate genetic information. The 16S rRNAs are commonly found in prokaryotic cells and have high content and copy number (80% of the total bacterial RNA).
There are nine conserved regions and nine highly variable regions in the 16S rRNA coding gene sequence. Among them, the V3-V4 region has good specificity and complete database information, which is the best choice for bacterial diversity analysis annotation (source).
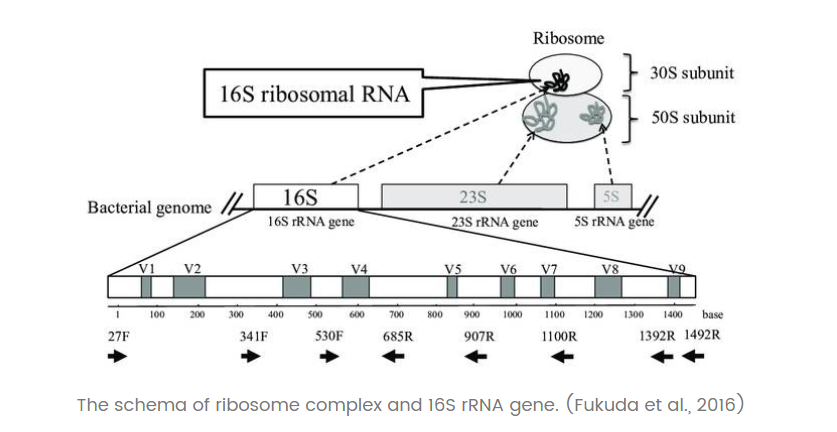
Q: What 's the application of amplicon sequencing technology ?
Amplicon sequencing finds extensive applications across various fields, enabling researchers to explore and understand microbial communities in diverse environments. Some notable application areas include:
(i) Medical Field: Amplicon sequencing allows for investigating the relationship between microorganisms and human diseases. It aids in exploring the role of microbial communities in conditions such as metabolic diseases, digestive diseases, autoimmune diseases, tumor cancer, neurological diseases, and other ailments. By profiling the microbiota, researchers can identify potential biomarkers, therapeutic targets, and interventions for improved human health.
(ii) Animal Husbandry: Amplicon sequencing plays a crucial role in studying the interactions between microbial communities in the gut, rumen, and other animal-associated environments. It helps researchers explore the impact of microbiota on animal reproduction, growth and development, nutritional health, immune function, and disease treatment. By understanding the microbial ecology in animal systems, scientists can optimize animal health and productivity.
(iii) Agriculture: Amplicon sequencing facilitates the investigation of microbial interactions within the rhizosphere and other plant-associated environments. Researchers can study the effects of agricultural practices such as tillage, fertilization, and crop rotation on soil microbial communities. Understanding the interplay between microorganisms and plants aids in improving agricultural productivity, nutrient cycling, plant health, and sustainable farming practices.
(iv) Environmental Studies: Amplicon sequencing allows for the characterization of microbial communities in various environments, including polluted settings like haze, dust, residential areas, shopping malls, and natural habitats such as water bodies and marine ecosystems. It aids in understanding the distribution, composition, and dynamics of microbial populations in different ecosystems. Additionally, amplicon sequencing contributes to research on organic fertilizer fermentation and treatment, sewage treatment, oil degradation, and other environmental processes.
(v) Bioenergy: Amplicon sequencing plays a vital role in bioenergy research by enabling the identification and characterization of special functional strains and genes relevant to biofuel production. It aids in gene mining and the development of engineered bacteria with enhanced capabilities for bioenergy production.
(vi) Special Extreme Environments: Amplicon sequencing is instrumental in studying microbial taxa thriving in extreme environmental conditions, such as extreme temperatures, high salinity, acidic or alkaline pH, and other challenging settings. By analyzing the microbial diversity and functional potential in these extreme environments, researchers gain insights into the adaptation strategies of microorganisms and their role in ecosystem dynamics (source).
Q: What are the types of samples for amplicon sequencing ?
Amplicon sequencing techniques, including 16S, 18S, and ITS amplicon sequencing, are applicable to a wide range of sample types (source).
Q: What should I do if the number of non-chimeric reads is low after the denoising step?
If you notice that the number of non-chimeric reads is low after the denoising step, you may need to adjust the --p-min-fold-parent-over-abundance parameter in the Q2FeatureInferencePE and Q2FeatureInferenceSE tasks.
This parameter specifies the minimum abundance required for potential parent sequences of a sequence being tested as chimeric. It is expressed as a fold-change relative to the abundance of the sequence under test. Values should be greater than or equal to 1 (i.e., potential parent sequences should be at least as abundant as the sequence being evaluated).
By default, the parameter is set to 1. It is recommended not to exceed a value of 16.
Q: What is the consequence of setting the --p-min-fold-parent-over-abundance parameter to a higher value?
Increasing this parameter generally leads to a higher number of non-chimeric reads being retained. As a result, the Shannon index tends to increase, since allowing more reads to pass through mechanically reveals a greater observed diversity.
Q: What artifacts could influence the Shannon Index?
Several factors can influence the Shannon index. This index can be calculated using different logarithmic bases, and each tool often uses a specific base by default. This variation explains why discrepancies may arise when comparing results across different tools or with published literature.
Other factors may also contribute to these variations, such as the sequencing technologies, the selected molecular targets, the animal species studied, the geographical location, as well as the transition from OTUs to ASVs.
For reference, ASVs are now preferred in most cases due to their higher resolution and reproducibility.
It is important to remember that the Shannon index is a relative measure. Its primary value lies in the differences observed between sample groups. This index is particularly sensitive to experiment-specific parameters.
Q: How can I minimize batch effects and ensure meaningful comparisons across studies after using --p-min-fold-parent-over-abundance parameter ?
To minimize batch effects and allow for meaningful comparisons between different studies, It's recommended ideally eprocessing all relevant datasets using the same parameter value. This value should be as parsimonious as possible; that is, the lowest value from your benchmark that still ensures a sufficient number of non-chimeric reads is retained.
Q: Why do I see weird quality check plots, and how should I assess read quality properly?
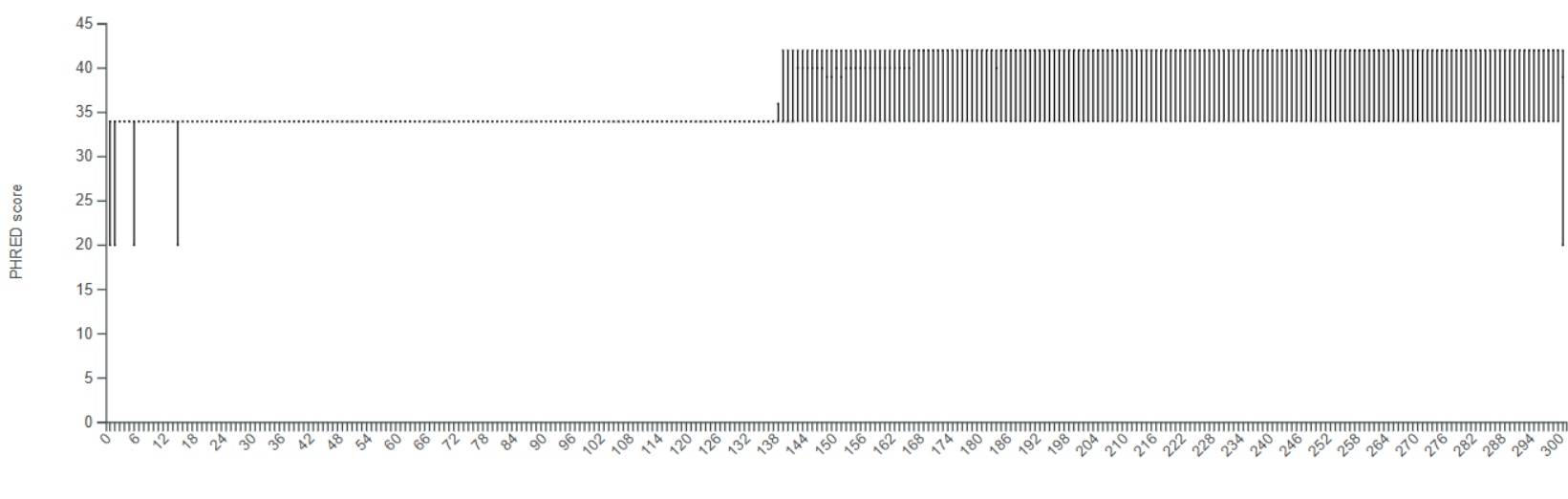
If your data comes from a NovaSeq or iSeq platform, it is normal to observe unusual quality check plots when using the Constellab pipeline.
To better assess the quality of your reads, we recommend using the FastQC and MultiQC tasks available in the gws_omix brick.
However, in all cases, you must also run the Q2QualityCheck task.
Q: what's the difference between Absolute Abundance vs Relative Abundance in the Microbiome
The main differences between absolute and relative abundance are as follows:
- Absolute abundance provides the actual count of microorganisms, which reflects the true number of microbes in the sample.
- Relative abundance describes the proportional relationship between different microorganisms within a sample, allowing for comparison of their relative distributions (source).
Q: When to Use Absolute Abundance and When to Use Relative Abundance?
Absolute abundance: If the goal is to determine the actual number of microorganisms (such as in disease monitoring or precise quantification of microbial load), absolute abundance is more reliable.
Relative abundance: If the focus is on understanding the community structure and comparing the proportions of different microorganisms within a sample (such as in ecological studies of microbial populations), relative abundance is often preferred. This approach highlights the proportional relationships among microbes within the community (source).
Q:Why LINDA AND DESEQ2 DETECT DIFFERENT KOs in16s Functional Analysis Prediction
Different statistical methods detecting different features is completely expected and normal. Here's why:
STATISTICAL DIFFERENCES:
- LinDA: Uses compositional data analysis, more conservative approach
- DESeq2: Uses negative binomial distribution, often detects more features
PRACTICAL IMPLICATIONS:
- Features detected by BOTH methods = HIGH CONFIDENCE results (focus on these)
- Features detected by ONE method = MODERATE CONFIDENCE (still valid)
- Each method has different sensitivity to low-abundance features
Let’s focus on analyzing this dataset:
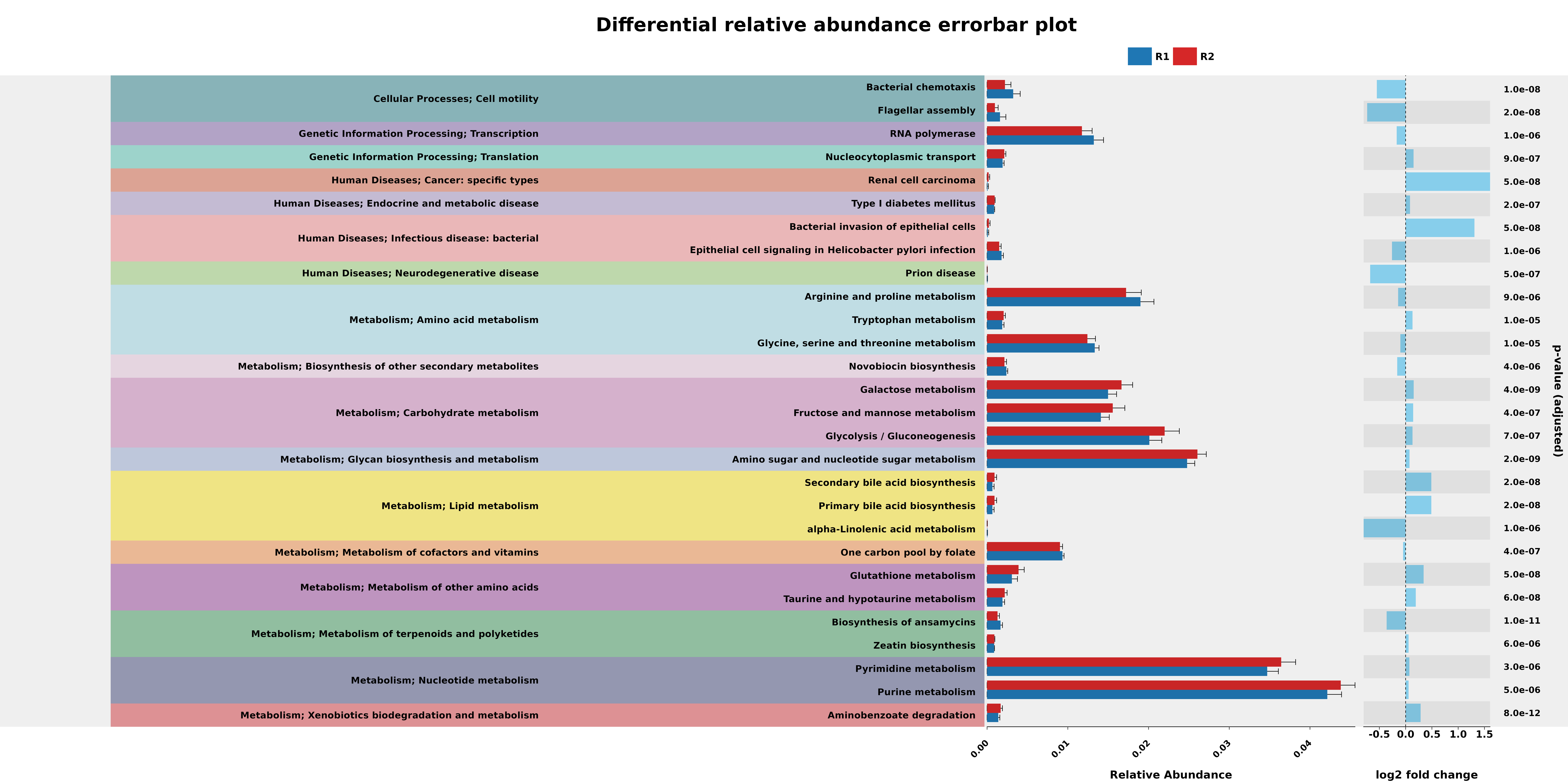
.png)
Overall statistics
- LinDA: 58 significant pathways
- DESeq2: 105 significant pathways (as expected, it’s more sensitive)
- Consensus: 55 pathways in common → 94.8% of LinDA results overlap with DESeq2
- This high overlap (55 shared pathways) suggests the findings are very robust.
Focus: ko00052 — Galactose metabolism
Status: Detected by both methods → high confidence
Your numbers:
- LinDA p-adjust: 0.030 (significant)
- DESeq2 p-adjust: 0.000000004 (highly significant)
- Log2 fold change: +0.152
- Pathway class: Metabolism → Carbohydrate metabolism
Interpretation:
- Galactose metabolism is enriched in R2 vs R1.
- The positive log2 fold change indicates higher activity in R2.
- Agreement between LinDA and DESeq2 strengthens confidence in this result.
- DESeq2 shows stronger significance, which fits its higher sensitivity.
Other high-confidence pathways (both methods)
- Primary bile acid biosynthesis (ko00120) — Log2FC: +0.493
- Secondary bile acid biosynthesis (ko00121) — Log2FC: +0.493
- Ubiquinone biosynthesis (ko00130) — Log2FC: +0.603
- Glutathione metabolism (ko00480) — Log2FC: +0.355
- Arginine and proline metabolism (ko00330) — Log2FC: −0.141
These point to core metabolic differences between R1 and R2.
What this means biologically ?
- Bile acid metabolism: Both primary and secondary pathways are enriched in R2
Consistent with shifts in gut microbiome composition
May reflect differences in dietary fat processing
- Carbohydrate metabolism: Multiple pathways (including galactose) are altered
Suggests distinct energy-use patterns between groups
- Antioxidant systems: Glutathione metabolism is enriched in R2
May indicate different oxidative stress responses
- Amino acid metabolism: Mixed directionality (some enriched, some depleted)
Points to broader metabolic reprogramming between groups
Recommendations
- The 94.8% overlap between LinDA and DESeq2 is excellent—treat the 55 consensus pathways as high-confidence results.
- For publication focus, highlight the bile acid pathways (ko00120, ko00121; Log2FC ~ 0.5) as strongest effects.
- Consider experimental validation of bile acid and glutathione pathways to confirm these computational findings.
