Introduction
Phylogenetics is the study of genetic relatedness of individuals of the same or different species, whether you're comparing individuals of the same species or tracking evolutionary changes across kingdoms. It’s a key tool in evolutionary biology, microbial genomics, and infectious disease research.
When you construct a phylogenetic tree, you’re building a visual model of those relationships. That tree may be rooted (if the common ancestor is known) or unrooted (if it’s not). The branches represent evolutionary paths, and the lengths often reflect evolutionary time or genetic distance. While every tree is ultimately an estimate, your choice of construction method can impact accuracy, speed, and interpretability.
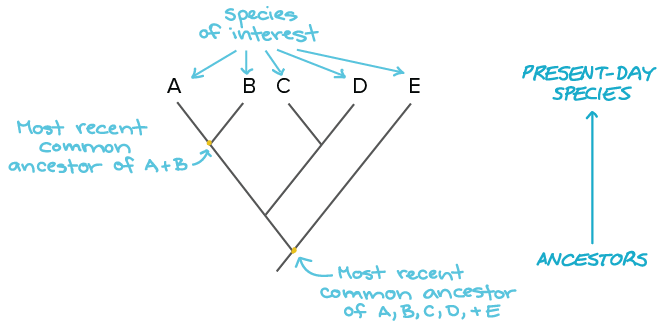
Let’s take a quick look at the most common approaches, such as maximum likelihood and Bayesian inference.
4 common phylogenetic tree construction methods
Distance-matrix methods
Distance-matrix methods are some of the fastest ways to construct a phylogenetic tree. After aligning your sequences with multiple sequence alignment software, the method calculates genetic distances (i.e., mismatches) and organizes them into a matrix.
From this, a tree is generated in which closely related sequences cluster under the same internal node. Two common techniques are:
- Neighbor Joining (NJ) builds unrooted trees without assuming equal evolutionary rates.
- UPGMA builds rooted trees but assumes a constant rate of evolution across all lineages, making it less popular for most real-world datasets.
Maximum parsimony
This method looks for the tree that requires the fewest changes, essentially offering the simplest evolutionary explanation. It evaluates every possible tree and selects the one with the least homoplasy (convergent evolution).
Simple doesn’t always mean better, though. Maximum parsimony isn’t statistically consistent and can miss complex evolutionary patterns.
Maximum likelihood
Maximum likelihood is the gold standard in phylogenetics. It evaluates the probability of your observed sequences under different tree topologies and chooses the one with the highest overall likelihood.
The upside: It’s powerful and detailed. The downside: It’s computationally demanding, especially for larger datasets.
It assumes each site evolves independently, calculating likelihoods at every bifurcation point. When more than four sequences are analyzed, sequence order can introduce bias, which can be solved by randomizing the process and selecting a consensus tree.
Bayesian inference
Bayesian phylogenetics builds on likelihood models by adding prior probabilities. It produces a range of trees, each with a posterior probability, giving you a clear sense of uncertainty and variation in your dataset.
It’s great for nuanced analysis and supports complex evolutionary models. Tools like MrBayes, BEAST, and RevBayes are often used for this approach.
Quick comparison: Phylogenetic tree construction methods
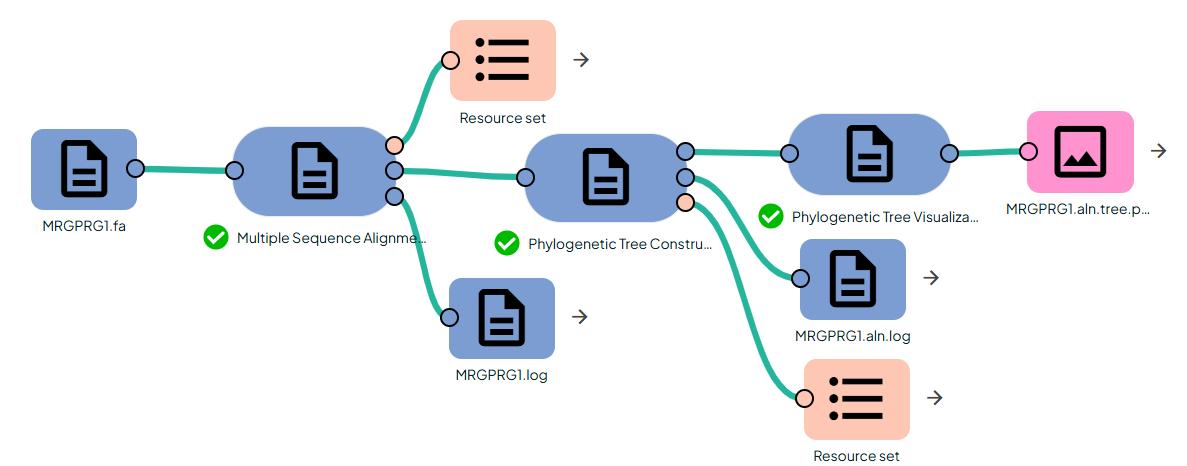
In Constellab this small pipeline builds and visualizes a phylogenetic tree from a set of homologous nucleotide or protein sequences.
It is composed of three tasks:
- Multiple Sequence Alignment (MSAVis)Uses MAFFT L-INS-i to produce a high-accuracy multiple alignment
- Phylogenetic Tree Inference (IQ-TREE)Wraps the iqtree binary to infer a maximum-likelihood tree from the aligned FASTA file, with optional ModelFinder Plus (MFP) model selection and ultrafast bootstrap support values.(https://github.com/iqtree/iqtree3)
- Phylogenetic Tree Visualization (PhyTreeViz)Uses phyTreeViz to render a clean PNG figure from the Newick tree (
.treefile), with optional display of branch lengths and bootstrap/confidence values.(https://github.com/moshi4/phyTreeViz)
The typical use case is: you start from raw sequences (FASTA), compute a high-quality alignment, estimate a maximum-likelihood tree with branch lengths and support values, and finally generate a figure that can go directly into a report or publication.
🧰 Prerequisites
- Access to Constellab and a valid Digital Lab environment
- Installed bricks: gws_omix ≥ 0.13.5
- Input file:FASTA file with ≥2 sequences (DNA or protein).This will be used by MSAVis and then propagated as an aligned FASTA to IQ-TREE.
🧪 Workflow: Step by Step
1. Multiple Sequence Alignment (MSAVis)
Task: Multiple Sequence Alignment
This task :
- Reads the input FASTA.
- Optionally truncates to at most 500 sequences (internally).
- Runs MAFFT L-INS-i if the input is not already aligned:
mafft --localpair --maxiterate 1000 --thread <threads> input.fa > output.aln.fa
- Detects whether sequences look like DNA or protein and selects a color scheme.
- Renders one or several PNG figures using pyMSAviz, automatically paginating long alignments.
2. Phylogenetic Tree Construction (IQ-TREE)
Task: Phylogenetic Tree Construction
This script calls the iqtree executable on the aligned FASTA:
By default:
-
-m MFP runs ModelFinder Plus to automatically select the best substitution model for your data and then infers the ML tree under that model. -
-bb 1000 runs ultrafast bootstrap with 1000 replicates to obtain branch support values. -
-nt 4 uses 4 CPU threads.
Inputs
-
in (path / File)Aligned FASTA from the MSA step (aligned_fasta output of MSAVis).
Configuration
-
prefix (string, default: tree)Base name for all IQ-TREE outputs:<prefix>.treefile<prefix>.log<prefix>.iqtreeand other IQ-TREE intermediate files. -
model (string, default: MFP)Substitution model string passed to -m. Examples:MFP – ModelFinder Plus (automatic model selection; recommended default). -
bootstrap (int, default: 1000)Number of ultrafast bootstrap replicates (-bb).0 → no bootstrap (fastest, but no support values).≥ 1000 recommended for robust supports. -
threads (int, default: 4)Number of threads passed to -nt.
Outputs
In the output directory:
-
<prefix>.treefileNewick tree with branch lengths and (if bootstraps were run) support values on internal nodes. -
<prefix>.logFull IQ-TREE log: details about model selection, tree search, convergence, etc. -
<prefix>.iqtreeSummary file with model parameters, log-likelihood, AIC/BIC, etc.
Additional files (e.g. .ckp.gz, .uniqueseq.phy) may also be created by IQ-TREE and remain in the same directory.
3. Phylogenetic Tree Visualization (PhyTreeViz)
Task: Phylogenetic Tree Visualization
This task uses the phyTreeViz Python library to turn the Newick tree into a publication-quality PNG.
The visualization:
- always uses branch lengths from the Newick to define the horizontal scale;
- can optionally display:branch lengths as numeric labels,bootstrap / confidence values (if present in the Newick, e.g. from IQ-TREE
-bb or -b).
Inputs
-
tree_file (File)Newick tree file, typically IQ-TREE’s <prefix>.treefile.
Configuration
-
prefix (string, default: tree filename stem)Base name for the output figure. The PNG will be named <prefix>.tree.png. -
fig_height (int, default: 3)Height per leaf node, scaled by 0.1 inside the task:Effective height = fig_height × 0.1.With 3, you get 0.3 per leaf, good for medium/large trees.Increase to add more vertical spacing between taxa. -
fig_width (int, default: 120)Figure width, also scaled by 0.1:Effective width = fig_width × 0.1.With 120, you get 12.0 units width.Increase for large trees or when branch lengths span a wide range. -
leaf_label_size (int, default: 8)Font size for leaf (tip) labels. -
align_leaf_label (bool, default: True)If True, aligns leaf labels vertically, producing a neat column on the right. -
show_branch_length (bool, default: False)If True, prints numerical branch lengths along the edges.On large trees this can become crowded; recommended for smaller trees or close-ups. -
show_confidence (bool, default: True)If True, renders node support / bootstrap values when available in the tree. -
dpi (int, default: 300)Output figure resolution.300 → print-quality, good general default.You can increase to 400–600 for high-resolution figures.
Outputs
-
tree_figure (File)A PNG figure named <prefix>.tree.png, containing:Rooted or unrooted tree layout as provided by IQ-TREE.Branch lengths mapped to the horizontal axis.Leaf labels aligned in a column (if enabled).Optional branch length text and bootstrap support values.A scale bar indicating the branch length unit.
🧩 Putting it all together
- Run Multiple Sequence AlignmentInput: raw FASTA sequences (
sequences_fasta).Output: aligned_fasta + one or more PNGs (msa_outputs).
- Run IQ-TREE wrapperInput:
aligned_fasta from MSAVis.Configure model = "MFP", bootstrap = 1000, threads as available.Output: <prefix>.treefile with branch lengths and bootstrap supports.
- Run PhyTreeVizInput: the
<prefix>.treefile from IQ-TREE as tree_file.Configure fig_height, fig_width, leaf_label_size, show_confidence = True.Output: <prefix>.tree.png, ready to be used in figures and reports.