Do you have genomic data and want to contextualize your metabolic model?
You’re in the right place!
Using differential gene expression (DGE) results, this pipeline allows you to integrate transcriptomic data into your metabolic model to generate a context-specific version of it.
Input Requirements
To start, you need to provide a table summarizing the differentially expressed genes (DEGs).
This table must include:
- A column with gene identifiers
- A column with the log2 fold change (log2FC)
The log2 fold change represents the difference in gene expression between a given condition and the control condition.
Once your DEG table is ready:
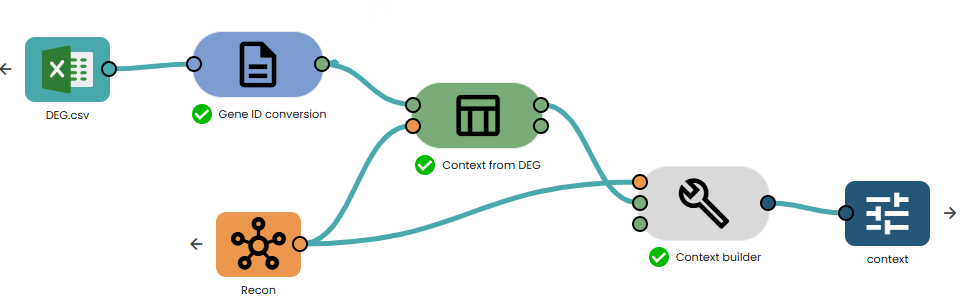
- Connect it to the task "Context from DEG".
- Fill in the required parameters.
- Run the pipeline.
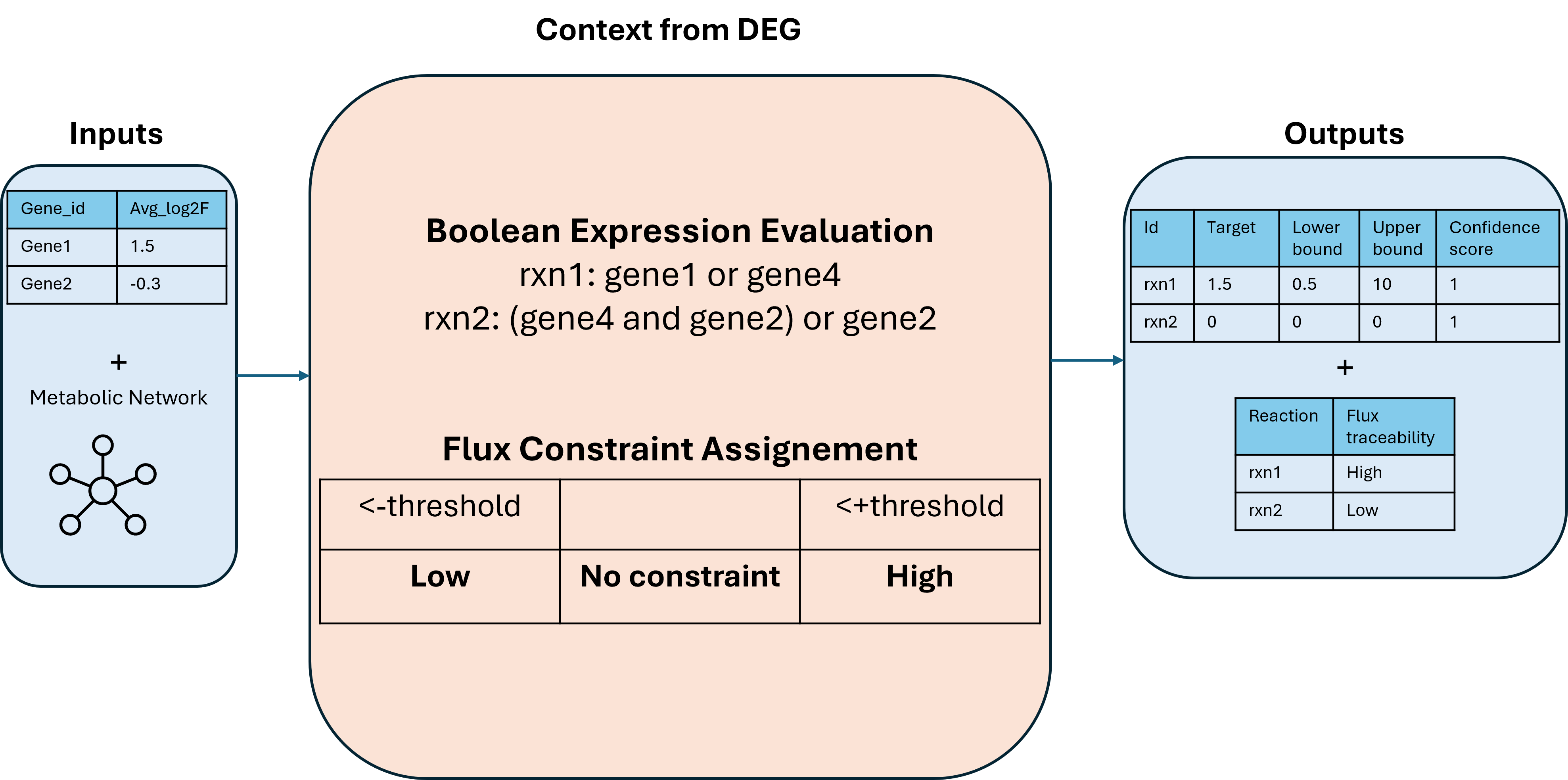
The output will be a context Table containing the reactions of your metabolic model and their associated flux bounds.
Then, you can connect it to the context builder Task. (see below)
How Does the Pipeline Work?
The contextualization pipeline is based on GPR rules (Gene–Protein–Reaction associations).
It operates under a key biological assumption:
If a gene is more expressed, its corresponding protein will be more abundant, leading to an increased flux through the associated reaction — and vice versa.
Here’s how it works step by step:
- Identify gene expression changes:
The pipeline reads your DEG table and detects which genes are overexpressed and which are underexpressed.
- Interpret GPR rules:
Each reaction in the model is linked to one or more genes via a GPR rule, which can look like:
gene1 OR gene2
This means the reaction can occur if either gene1 or gene2 is expressed.
- Resolve the GPR logic:
The pipeline evaluates each GPR rule based on the expression status of the genes.
If the rule evaluates to true, the reaction remains active (non-zero flux).
If the rule evaluates to false, the reaction is knocked out.
- Set reaction bounds and targets:
Depending on the outcome of each GPR evaluation, reaction bounds are adjusted to reflect the condition-specific metabolic state.
The result is a context table that contains:
- The list of reactions in the model
- Their updated flux bounds based on gene expression data
This context file can then be used in downstream analyses, such as digital twin simulations or condition-specific metabolic modeling.
Hypothesis
The following hypothesis is assumed in the pipeline:
- The first is the GPR rule. See the previous part for more details.
- Another rule followed is that, to resolve the GPR Boolean rule, we use the fold change values. Thus, if it is an 'or' relationship, we take the maximum fold change between the two genes. If it's an 'and' relationship, we take the minimum fold change between the two genes.
