Introduction
STAR, an acronym for "Spliced Transcripts Alignment to a Reference," is a powerful and widely used bioinformatics tool for genome indexing and alignment. With its ability to handle both fasta and gtf files. STAR facilitates efficient and accurate mapping of high-throughput sequencing data to a reference genome.
It requires both a Fasta file containing the nucleotide sequences of the genome and GTF file providing information about gene annotation and genomic features. By combining these files, STAR constructs an index that allows for rapid identification of potential alignments between the sequencing reads and the reference genome.
Main functions of STAR index
STAR index using STAR permits to build a comprehensive index that enables efficient alignment of RNA-seq and scRNA-seq
reads to the reference genome. This indexing process greatly enhances the speed and accuracy of read alignment.
In this context STAR index is also used by STAR solo to generate raw matrix from scRNA-seq fastq files.
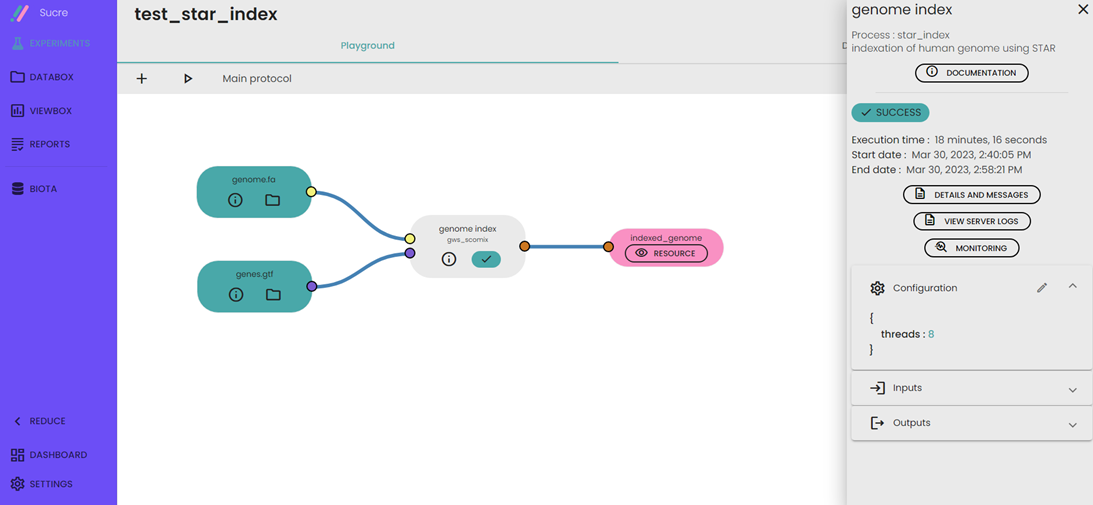
Steps to follow
- Ensure that the version 0.1.2 of gws_scomix brick is loaded
- So first, upload your fastqc folder , FASTA and GTF files to the Databox.
- Then, create a new experiment.
- Import your resource
- Link it to the task "Building a genome index" available in the brick gws_scomix.
- Specify number of threads
- Run your experiment
- A folder containing the indexed genome will be generated.

Comments (0)
Write a comment