Introduction
STARsolo (v 2.7.9a ) is a turnkey solution for analyzing droplet single cell RNA sequencing data (e.g. 10X Genomics Chromium System) built directly into STAR code. STARsolo output is designed to be a drop-in replacement for 10X CellRanger gene quantification output. It follows CellRanger logic for cell barcode whitelisting and UMI deduplication, and produces nearly identical gene counts in the same format. At the same time STARsolo is ~10 times faster than the CellRanger.
Main functions of STAR solo :
STARsolo inputs the raw FASTQ reads files, and performs the following operations :
- Error correction and demultiplexing of cell barcodes using user-input whitelist
- Mapping the reads to the reference genome using the standard STAR spliced read alignment algorithm
- Error correction and collapsing (deduplication) of Unique Molecular Identifiers (UMIa)
- Quantification of per-cell gene expression by counting the number of reads per gene
- Quantification of other transcriptomic features: splice junctions; pre-mRNA; spliced/unspliced reads similar to Velocyto
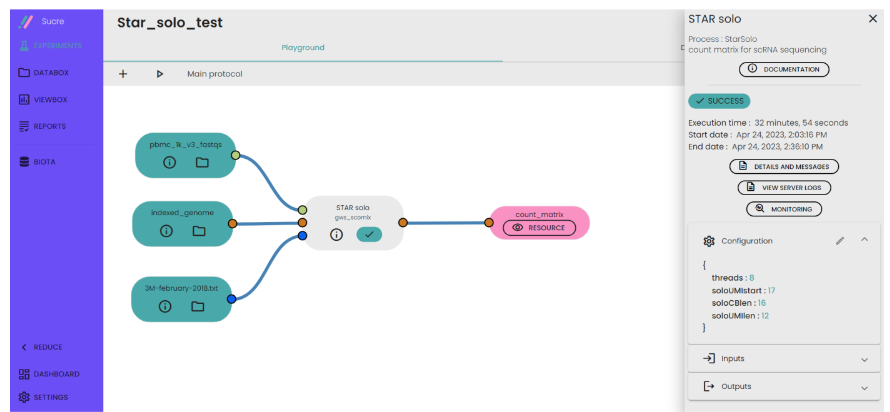
Steps to follow
- Ensure that the version 0.1.2 brick is loaded
- Upload your fastq folder , the right version of whitelist file that correspond to the kit used during the experiment to the Databox.
- Then, create a new experiment.
- Add the indexed reference genome generated by task "Building a genome index" available in the brick gws_scomix , fastq folder and whitelist file.
- Specify some parameters such as cell barcode length , UMI coding start and length.
- Run your experiment.
- Generate a folder containing the result of your experiment.
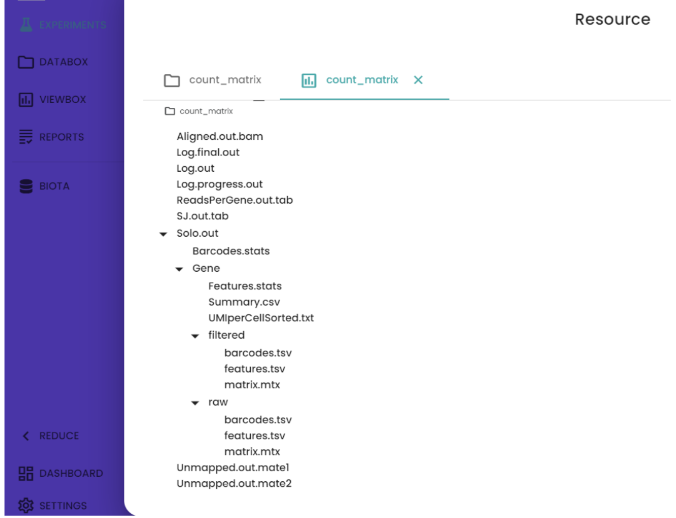
Description of output file
STAR solo generates almost the same files as Cell ranger. Both filtered and raw counts are generated. “Barcodes.tsv” contains the list of unique barcodes associated with each cell in the scRNA-seq dataset. “Features.tsv” contains the list of gene names and “matrix.mtx” stores information about gene expression levels associated with each gene and cell barcode combination.

Comments (0)
Write a comment