Introduction
PyDESeq2, a Python implementation of the DESeq2 method originally developed in R (click here) , is a versatile tool for conducting differential expression analysis (DEA) with bulk RNA-seq data. This powerful library is designed to cater to the diverse needs of Python users engaged in RNA-seq experiments.
Notably, PyDESeq2 excels in handling categorical factors within the context of DEA.
Categorical factors, such as experimental conditions are crucial components of the experimental design, representing distinct categories or groups that contribute to the variation in gene expression.
PyDESeq2's capability to effectively analyze and interpret differential expression across these discrete categories empowers researchers. It offers valuable insights into gene expression changes in response to different factors by unraveling the molecular intricacies associated with experimental conditions.
By implementing Wald tests, PyDESeq2 enables users to statistically evaluate the significance of these expression differences, providing a robust framework for unraveling the nuanced relationships between genes in RNA-seq studies.
To test PyDESeq2, a dataset extracted from the article titled "Senescence in Vascular Smooth Muscle Cells and Atherosclerosis" was utilized. This dataset contains samples comprising normal human cell controls and replicative senescence cells, sourced from the NCBI accession GSE171663.
Prerequesites
Steps to follow
I. Differential analysis:
- Import your gene expression matrix and metadata file in Constellab.
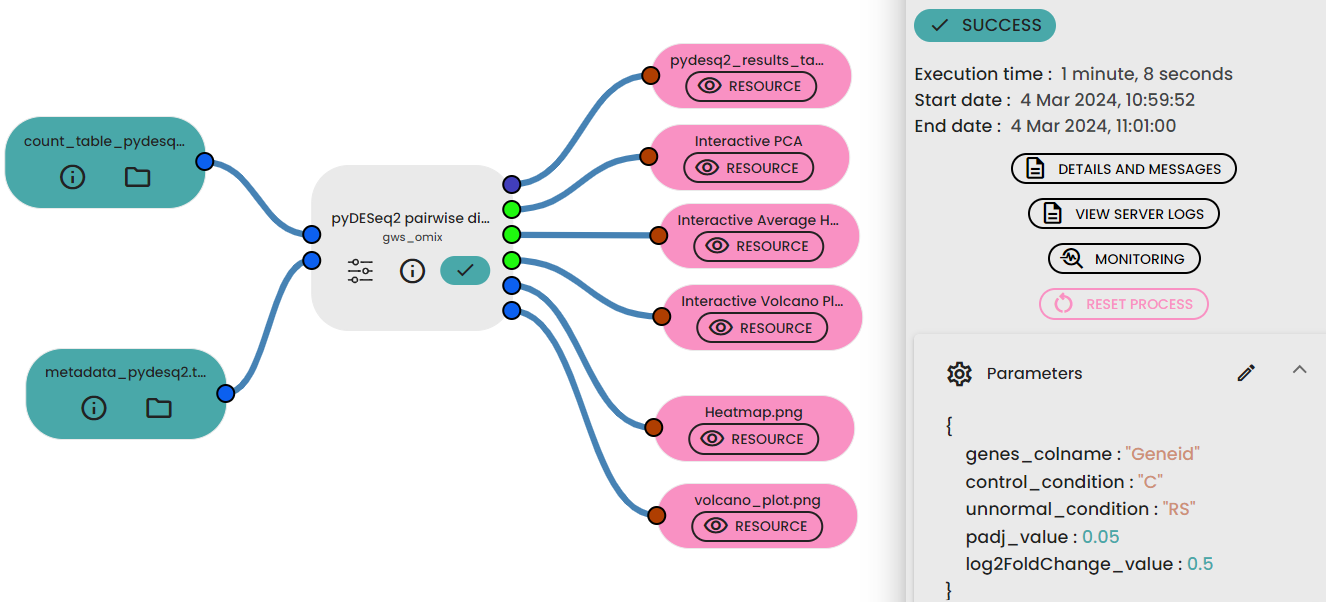
- Link it to the Task "pyDESeq2 pairwise differential analysis".
- Fill parameters : you must indicate Column name containing gene ids in expression matrix , normal_condition , unnormal_condition , padj_value and log2FoldChange value.
- Run your experiment.
II. KEGG Visualisation :
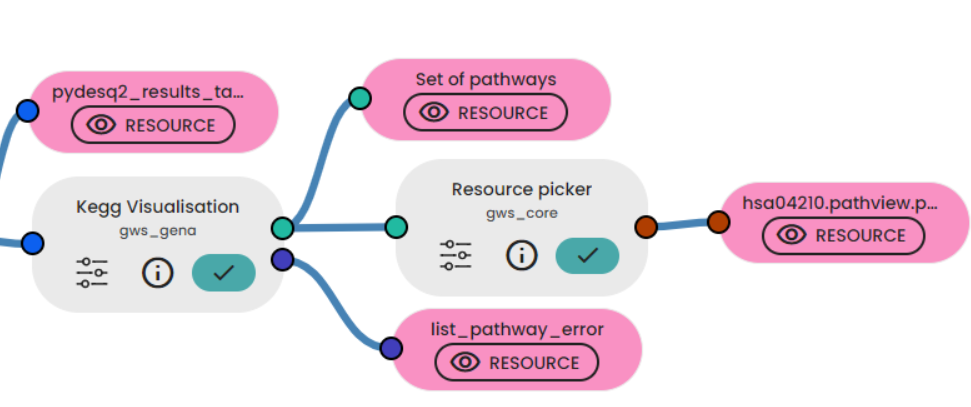
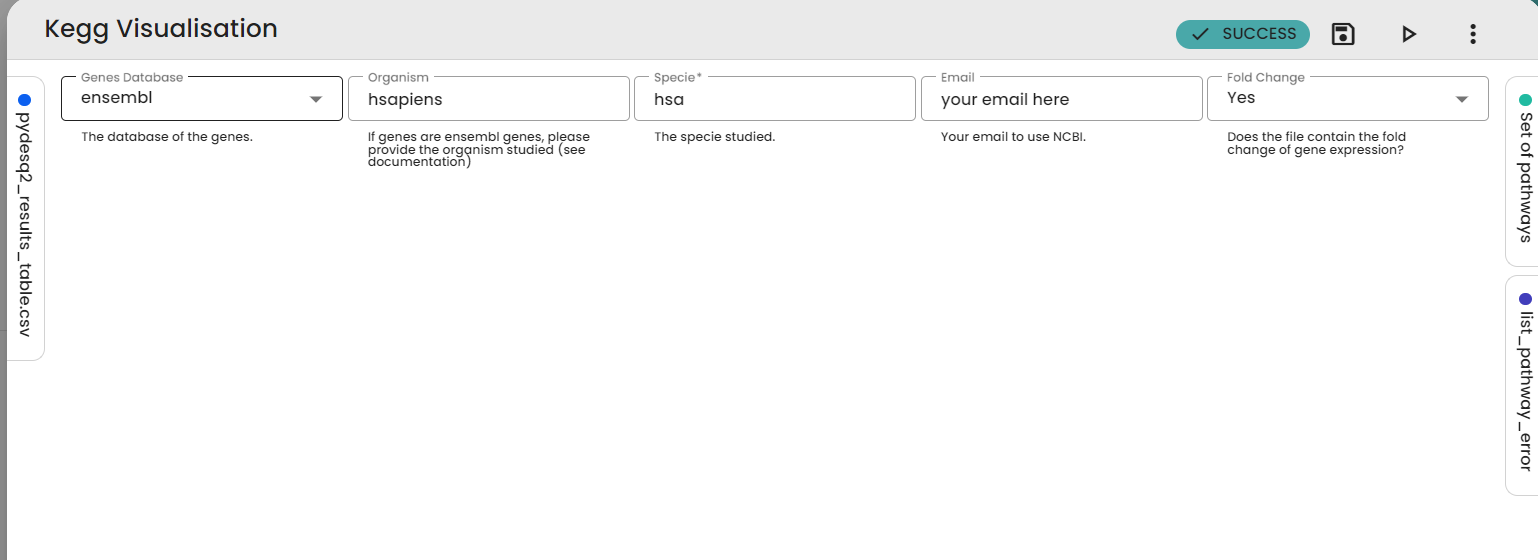
With the pydeseq2 table we have obtained, we can use the KEGG visualisation task to see the consequences of differential gene expression on pathways.
In the article they talk a lot about apoptosis, so we will extract this pathway (hsa04210).
Results
I. Differential analysis:
The experiment generates six outputs respectively:
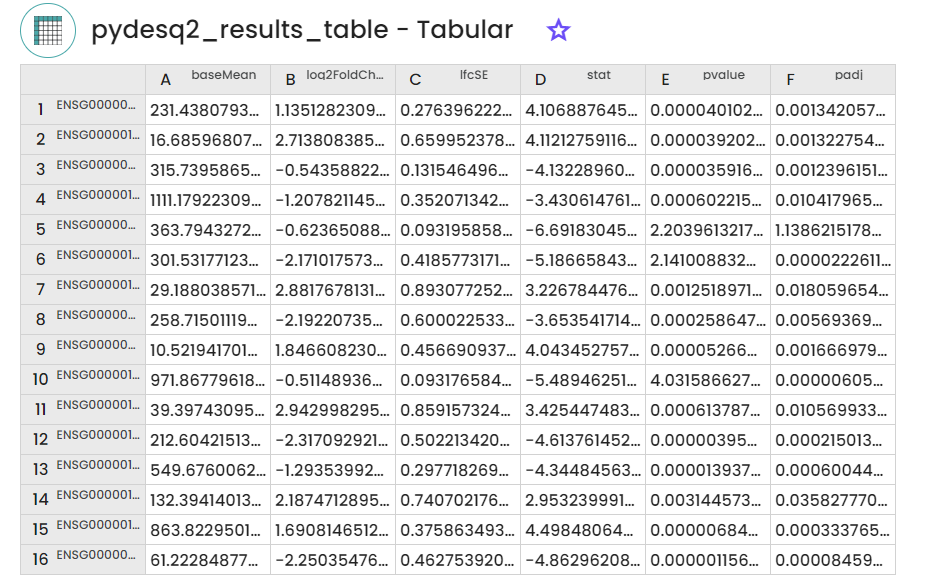
- A table containing differential expression results ,providing a summary of genes showing significant changes in expression levels between two conditions.
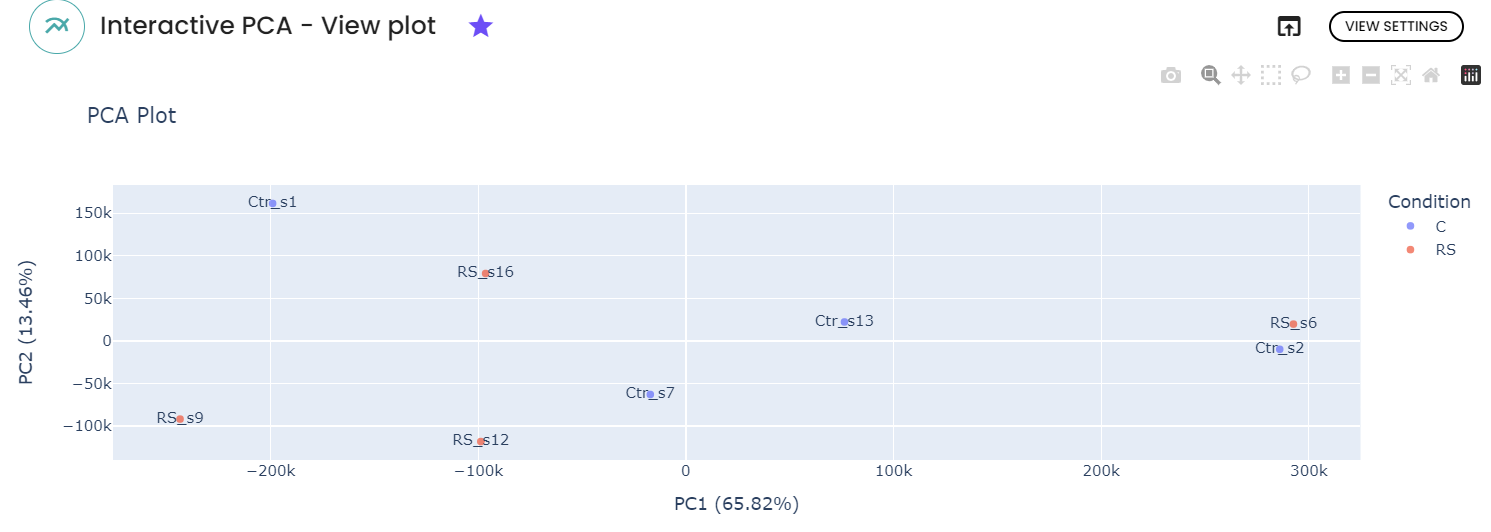
- A principal component analysis plot: allowing to explore the relationships between samples based on their gene expression profiles.
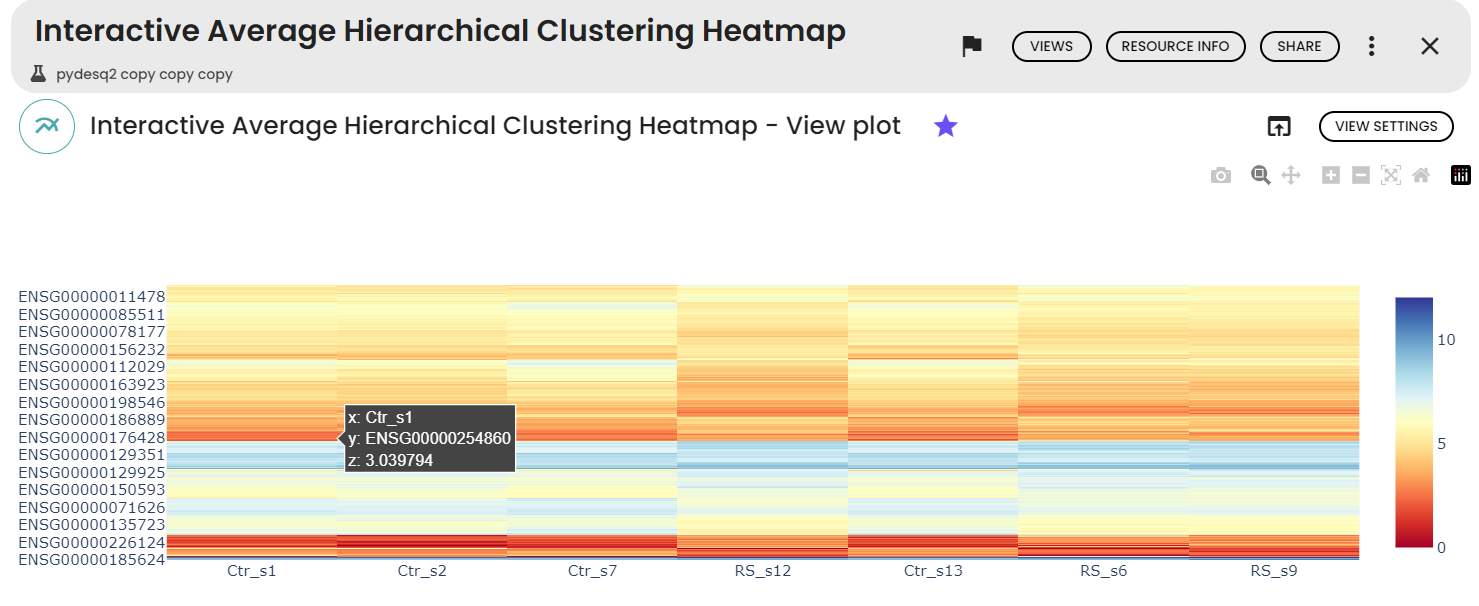
- An interactive average hierarchical clustering heatmap : the average linkage method and the Euclidean distance metric was used for hierarchical clustering. The clustering results to visually group similar expressed genes.
- A static hierarchical clustering heatmap : displaying average expression levels across different groups with rows representing individual genes ensembl id and columns representing samples. The hierarchical clustering dendrograms are typically displayed on the side of the heatmap, showing the relationships between samples based on their similarity in expression profiles.
- An interactive volcano plot and static volcano plot : It displays statistical significance ( represented as p-values) on the y-axis against fold change values on the x-axis for each feature (genes) in a dataset.
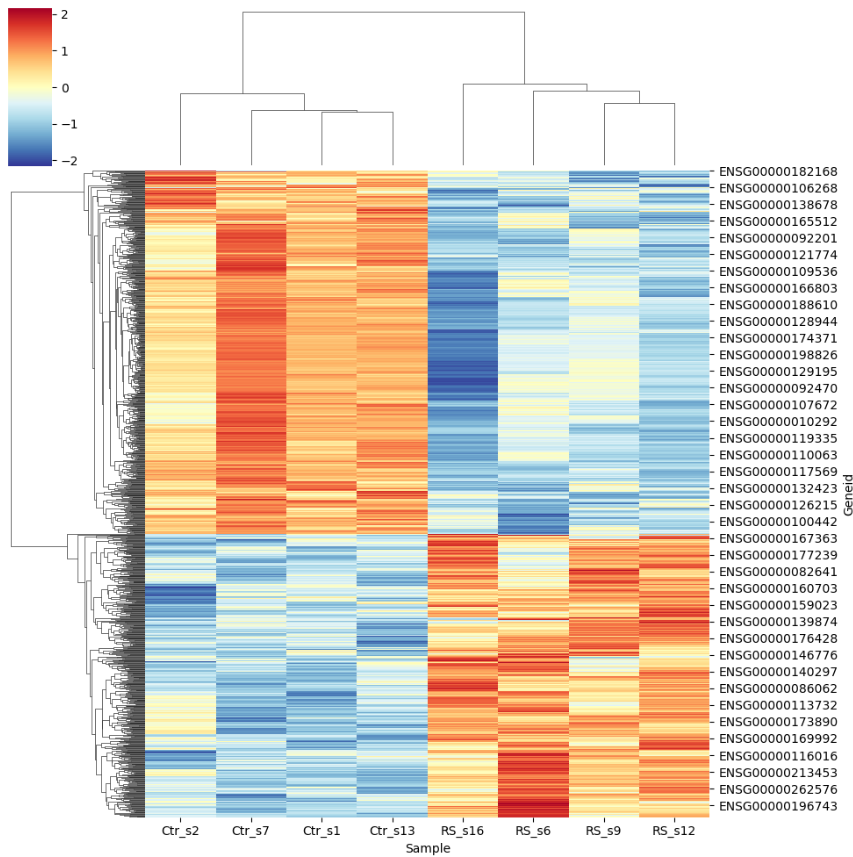
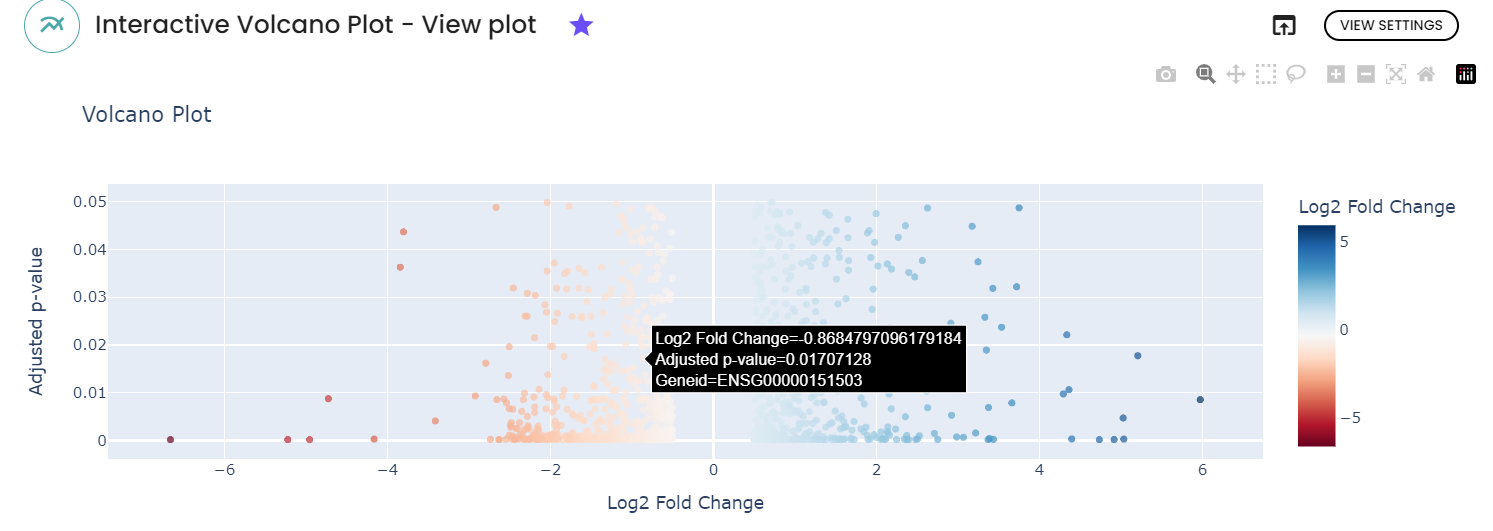
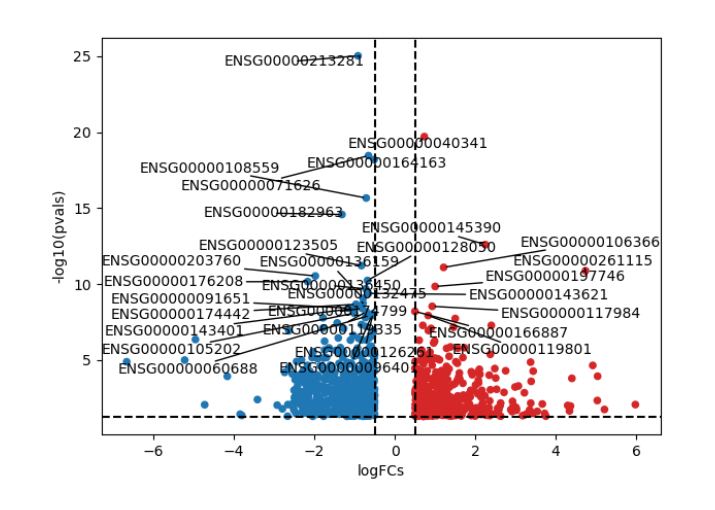
II. KEGG Visualisation :
In the article they talk a lot about apoptosis, so we will extract this pathway (hsa04210).
You can see that a lot of genes are mapped to this pathway. So the natural cycle of apoptosis is altered.
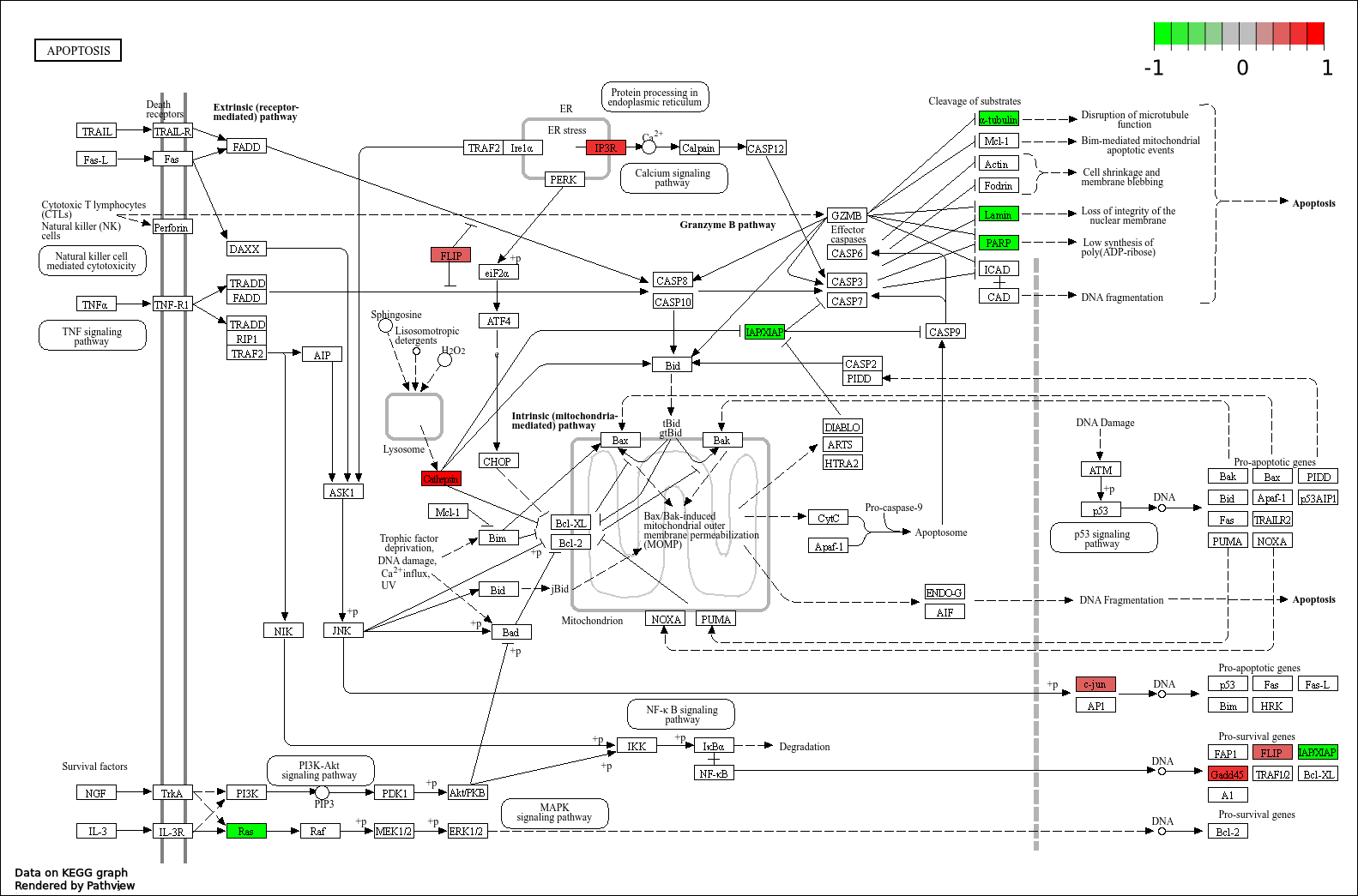
Conclusion
we observed that the results are similar to the original article namely the enrichment of the apoptosis pathway despite the use of different tool.

Comments (0)
Write a comment