Introduction
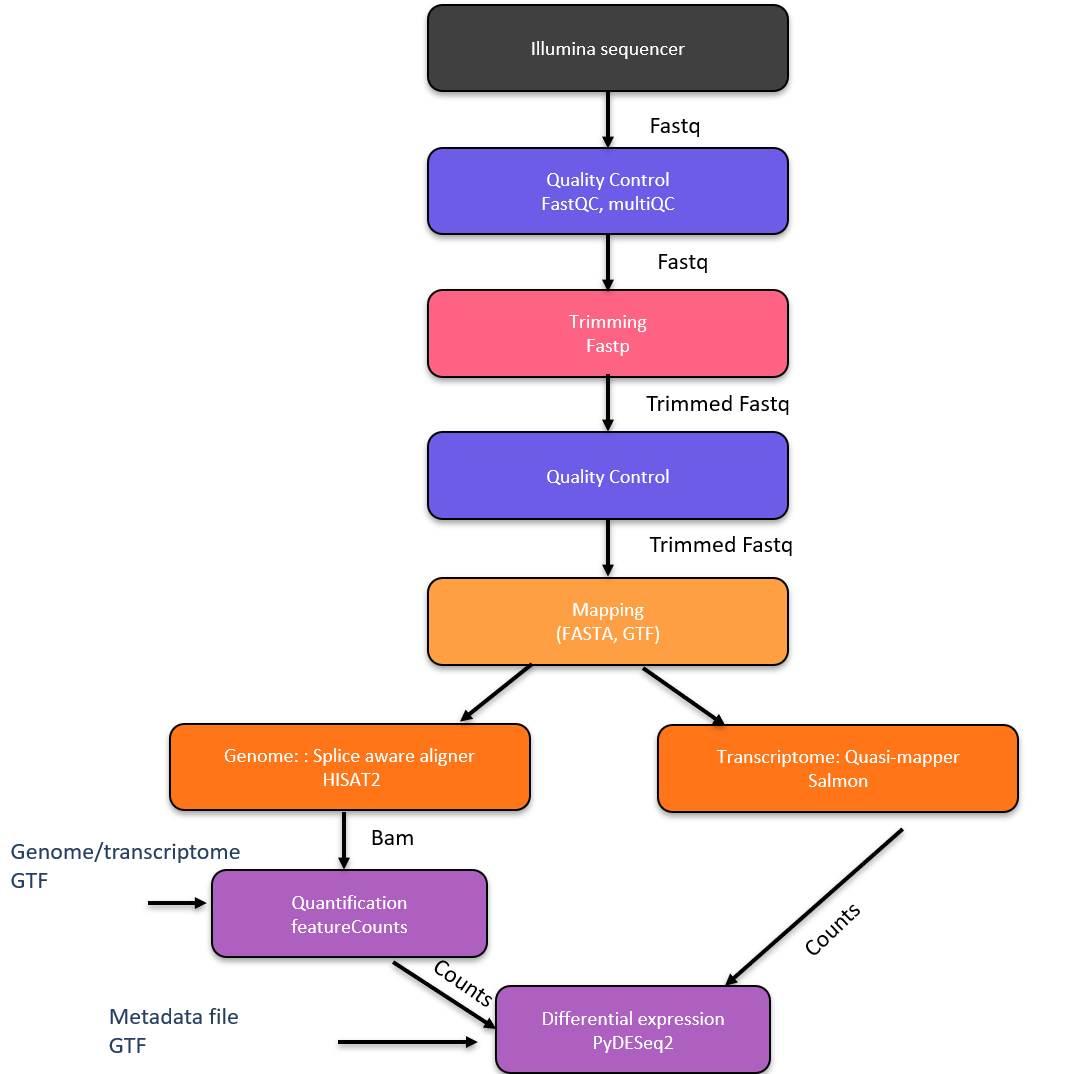
RNA sequencing (RNA-Seq) is used to study the transcriptome of living organisms. This high-throughput sequencing technique allows to measure the expression of an organism's genes and thus to compare the transcriptome of individuals under different conditions, to compare gene expression between different organs, at different times, etc.
Another major advantage of this technology is its ability to discover new isophorms, alleles, mutations (SNPs, InDels) within the sequenced genes. RNA-Seq is also an excellent tool for refining the annotation of assembled genomes.
This documentation presents the main steps you need to use RNA-Seq mapping.
🧰 Prerequisites
- Access to Constellab and a valid Digital Lab environment
- Installed bricks: gws_omix ≥ 0.13.10
Data upload and preparation
Input folder
One must upload one folder with all the sequencing data in the Databox. You must select the following format: Fastq folder.
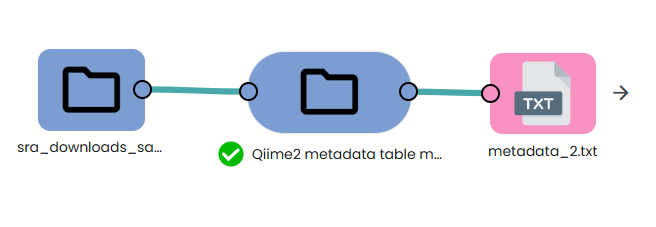
Making the ready-to-use metadata file
The uBiome Qiime2metadata table maker task automatically generates a ready-to-use metadata file when given a fastq folder as input. Once the metadata file is generated, you can add specific metadata columns in the expected file format (see below).
Example :
#author: Paulson, Robert
#data: 1996/08/17
#project: Chaos
#types_allowed:categorical or numeric
#column-type categorical categorical categorical
Sample forward-absolute-filepath reverse-absolute-filepath Timepoint Condition Group
t1_control-1 t1_control-1_1.fastq.gz t1_control-1_2.fastq.gz t1 control Female
t1_control-2 t1_control-2_1.fastq.gz t1_control-2_2.fastq.gz t1 control male
t1_rapid-1 t1_rapid-1_1.fastq.gz t1_rapid-1_2.fastq.gz t1 rapid Female
t1_rapid-2 t1_rapid-2_1.fastq.gz t1_rapid-2_2.fastq.gz t1 rapid Female
t1_rapid-3 t1_rapid-3_1.fastq.gz t1_rapid-3_2.fastq.gz t1 rapid male
t1_slow-1 t1_slow-1_1.fastq.gz t1_slow-1_2.fastq.gz t1 slow Female
t1_slow-2 t1_slow-2_1.fastq.gz t1_slow-2_2.fastq.gz t1 slow male
t1_slow-3 t1_slow-3_1.fastq.gz t1_slow-3_2.fastq.gz t1 slow male
t2_control-1 t2_control-1_1.fastq.gz t2_control-1_2.fastq.gz t2 control Female
t2_control-2 t2_control-2_1.fastq.gz t2_control-2_2.fastq.gz t2 control male
t2_rapid-1 t2_rapid-1_1.fastq.gz t2_rapid-1_2.fastq.gz t2 rapid Female
t2_rapid-2 t2_rapid-2_1.fastq.gz t2_rapid-2_2.fastq.gz t2 rapid male
t2_rapid-3 t2_rapid-3_1.fastq.gz t2_rapid-3_2.fastq.gz t2 rapid male
t2_slow-1 t2_slow-1_1.fastq.gz t2_slow-1_2.fastq.gz t2 slow Female
t2_slow-2 t2_slow-2_1.fastq.gz t2_slow-2_2.fastq.gz t2 slow Female
t2_slow-3 t2_slow-3_1.fastq.gz t2_slow-3_2.fastq.gz t2 slow male
t3_control-1 t3_control-1_1.fastq.gz t3_control-1_2.fastq.gz t3 control Female
t3_control-2 t3_control-2_1.fastq.gz t3_control-2_2.fastq.gz t3 control male
t3_rapid-1 t3_rapid-1_1.fastq.gz t3_rapid-1_2.fastq.gz t3 rapid Female
t3_rapid-2 t3_rapid-2_1.fastq.gz t3_rapid-2_2.fastq.gz t3 rapid male
t3_rapid-3 t3_rapid-3_1.fastq.gz t3_rapid-3_2.fastq.gz t3 rapid male
t3_slow-1 t3_slow-1_1.fastq.gz t3_slow-1_2.fastq.gz t3 slow male
t3_slow-2 t3_slow-2_1.fastq.gz t3_slow-2_2.fastq.gz t3 slow male
t3_slow-3 t3_slow-3_1.fastq.gz t3_slow-3_2.fastq.gz t3 slow Female
Protocol
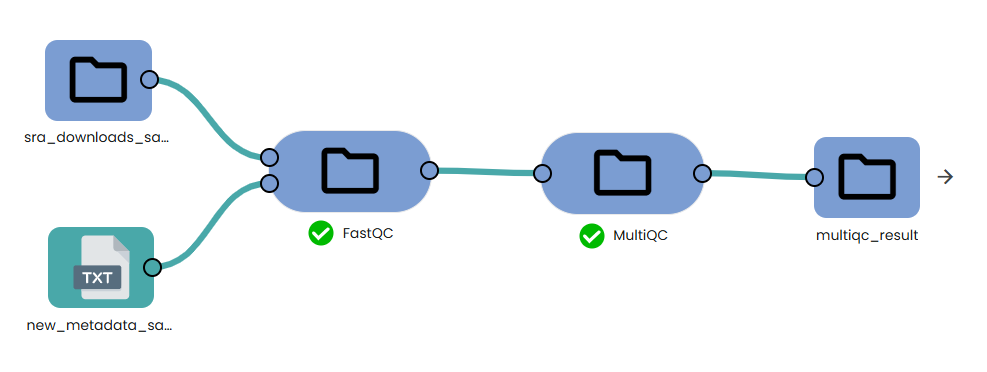
STEP 0 - Reads quality check OmiX – FastQC and MultiQC
This step is not mandatory. This task (task: OmiX - FastQC and MultiQC) allows to investigate visually sequencing quality from a sequencing dataset project.
Running FastQC is optional but highly recommended to visualise base-quality profiles, adapter content and sequence duplication. MultiQC aggregates all individual reports into a single html file.
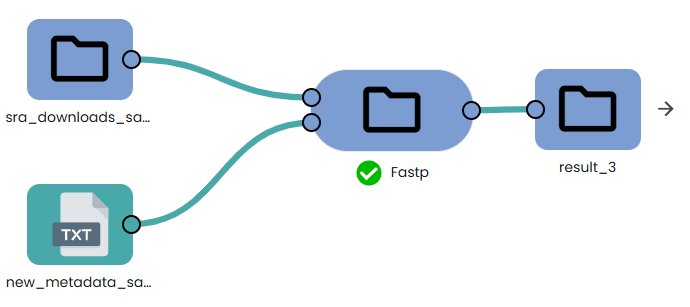
STEP 1 - Trimming
This step (task: OmiX - Fastp) is a lightweight wrapper around fastp, a fast all-in-one read-cleaning tool for FASTQ data.
In a single pass it
- detects and removes sequencing adapters,
- hard-trims a fixed number of bases from the 5′ end (optional),
- filters low-quality leading bases
STEP 2.a - Transcriptome approach
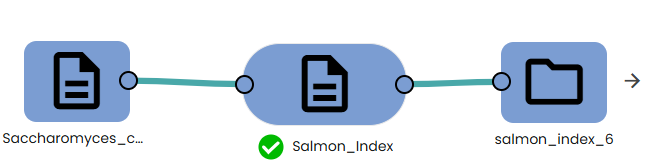
- Indexing:
To perform transcriptome mapping you first need to index your transcriptome data (task: OmiX - Salmon_Index).
We advise you to use transcriptome from reference databases (ensembl, NCBI, EBI...) which will offer you the annotation file of these genomes/transcriptome (i.e., the position of the genes in this genome. To get direct download files go to Ensembl website :
- Ensembl and choose your species, select cds option, then download the .fa , and gtf.gz file.
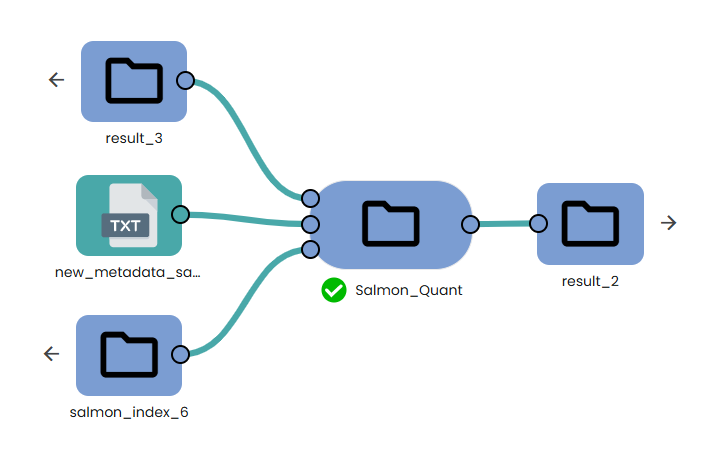
- Mapping and Gene transcription quantification:
Trimmed sequencing datasets are mapped (task: OmiX - Salmon_Quant) on the previously indexed transcriptome.
Metadata file must be provided to perform this step.
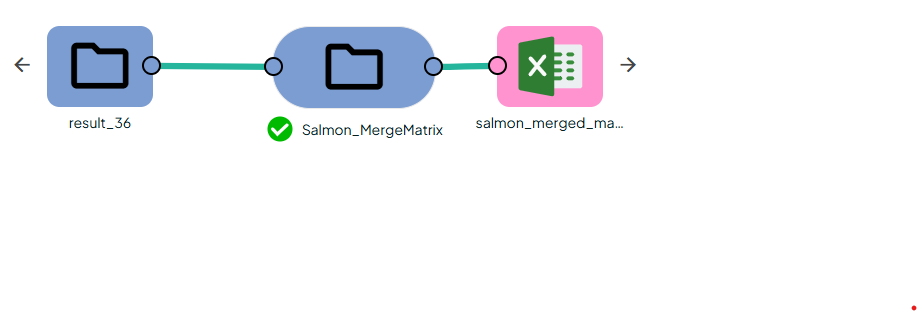
- Merging:
After salmon quant each sample owns a quant.sf file that holds fragment counts at the transcript level.
The task: OmiX - Salmon_MergeMatrix reads every quant.sf in a Salmon-Quant output folder, links each transcript to its parent gene using a supplied GTF, collapses the counts, and merges all samples into one tidy matrix ready for differential-expression analysis.
Information :
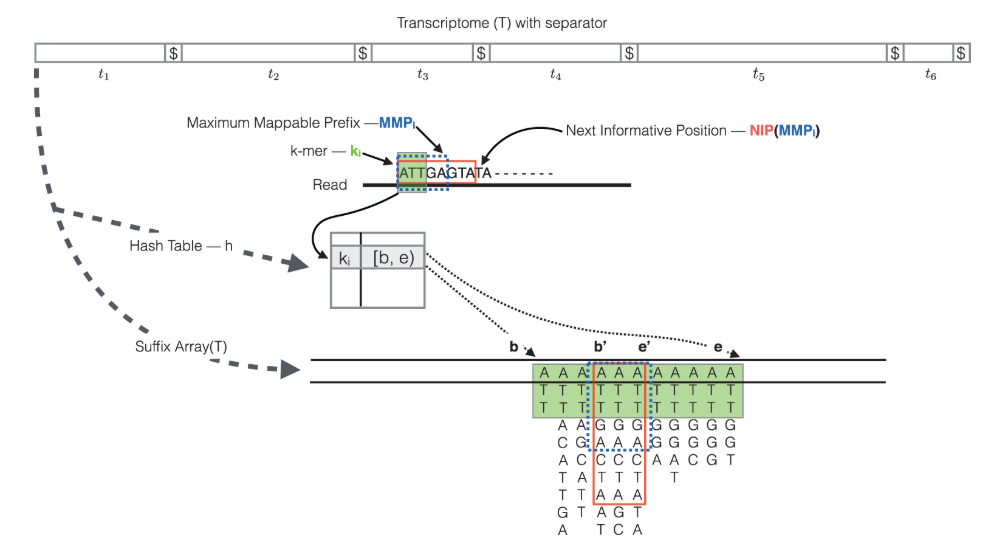
STEP 2.b - Genome approach
- Indexing:
To perform genome-based mapping using HISAT2, you must first build an index of your reference genome using the task: OmiX - Hisat2_Index task.
We advise you to use genome from reference databases (ensembl, NCBI, EBI...) which will offer you the annotation file of these genomes/transcriptome (i.e., the position of the genes in this genome. To get direct download files go to Ensembl website :
- Ensembl and choose your species, select dna option, then download the .fa and then merge them into a single file, then you have to download the gtf.gz file and decompress it.
🪄 Golden rules
- Match the version of your annotation (GTF/GFF)
Always use the same genome build and release (e.g GRCh38, GRCm39…).
- Use the main (primary) assembly, unmasked: Prefer the primary assembly . It contains the standard chromosomes without repeats or alternative scaffolds.
- → Avoid masked genomes (_rm, _sm).

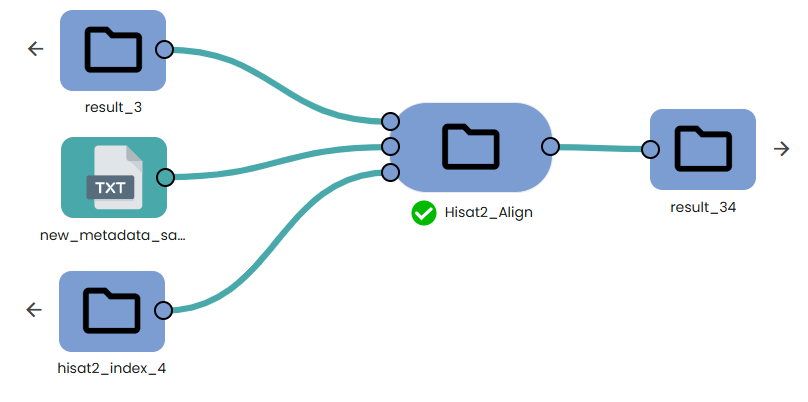
- Mapping
This task (task: OmiX - Hisat2_Align) wraps HISAT2 (for alignment) and samtools sort (for BAM creation).
It takes the trimmed FASTQ files, maps them to a pre-built HISAT2 genome index, and produces one coordinate-sorted BAM per sample.
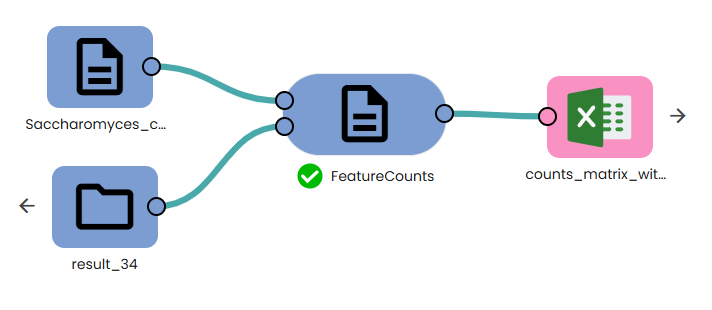
- Mapping and Gene transcription quantification:
This task (task: OmiX - FeatureCounts) runs featureCounts on all BAMs to count exon-aligned reads per gene (respecting strand, paired-end, and multi-mapping settings).
Parameters:
Information :
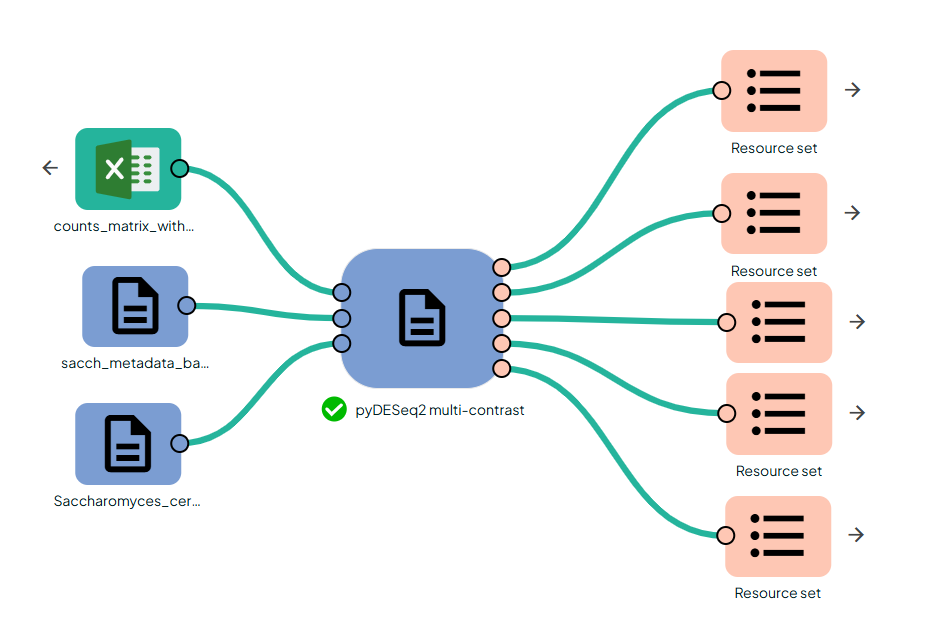
STEP 3 - Gene expression differential analysis
This task (task: OmiX - pyDESeq2 multi-contrast) performs automated differential expression analysis using the Python port of DESeq2 (
PyDESeq2). For every treatment level listed in the metadata it runs a separate Wald test against the chosen control, and—when two or more treatments are present—adds one pooled comparison “ALL vs CTRL.”
The design formula is built dynamically as: ~ <covariates> + Condition, where <covariates> includes any of timepoint_column, group_column, and any columns listed in extra_covariates provided they exist in the metadata. If no covariate column is available, the design is ~ Condition.
Each contrast yields a results table (pydesq2_results_table.csv), an interactive volcano plot, and a heatmap of the top 50 genes; a global VST-based PCA plot is produced as well. Together, these outputs give a complete statistical and visual summary of differential expression across all conditions in a single run.
Sample Timepoint Condition Group
t1_control-1 t1 control Female
t1_control-2 t1 control male
t1_rapid-1 t1 rapid Female
t1_rapid-2 t1 rapid Female
t1_rapid-3 t1 rapid male
t1_slow-1 t1 slow Female
t1_slow-2 t1 slow male
t1_slow-3 t1 slow male
t2_control-1 t2 control Female
t2_control-2 t2 control male
t2_rapid-1 t2 rapid Female
t2_rapid-2 t2 rapid male
t2_rapid-3 t2 rapid male
t2_slow-1 t2 slow Female
t2_slow-2 t2 slow Female
t2_slow-3 t2 slow male
t3_control-1 t3 control Female
t3_control-2 t3 control male
t3_rapid-1 t3 rapid Female
t3_rapid-2 t3 rapid male
t3_rapid-3 t3 rapid male
t3_slow-1 t3 slow male
t3_slow-2 t3 slow male
t3_slow-3 t3 slow Female
Parameters:
What each column means in this context