Introduction
Gene knockout simulation involves computational modeling techniques to predict the effects of gene deletion or inactivation within biological systems. By selectively disabling specific genes, researchers can simulate the consequences on cellular or organismal behavior, providing valuable insights into gene functions, regulatory pathways, and potential therapeutic targets. These simulations typically utilize mathematical models, such as constraint-based metabolic models or Boolean networks, to simulate the altered cellular phenotype resulting from gene knockout. Through these simulations, researchers can explore how gene knockout impacts cellular metabolism, signaling pathways, and other biological processes, aiding in the identification of essential genes, understanding disease mechanisms, and designing strategies for genetic engineering or drug discovery. Gene knockout simulations play a crucial role in systems biology, providing a powerful tool for hypothesis generation, experimental design, and advancing our understanding of complex biological systems.
In Constellab you can simulation gene knockout simulations, using the KOA Task; which we will see in more detail in this documentation.
Data upload
You need to upload your metabolic network with a context, if necessary. Here, we will use the E.coli core model with the lower bounds of the EX_o2_e reaction at 0 and the EX_glc__D_e reaction at -20.
You also need a file to import the reactions you want to knock out. Here we upload this table:
Protocol
Once these files have been imported, all you need to do is connect the various Tasks as shown below:
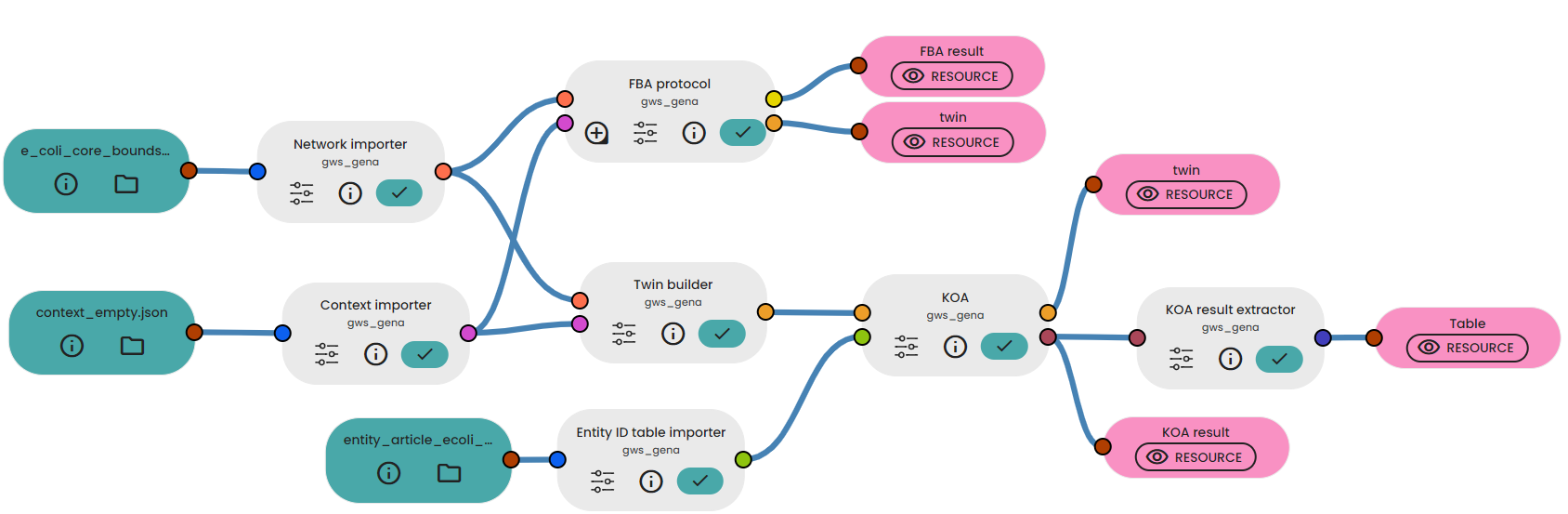
For the parameters of the FBA protocol and the KOA Task, set the optimisation of the biomass to "maximize". For the KOA Result Extractor Task we want to extract the flux "network_BIOMASS_Ecoli_core_w_GAM".
Results
The estimated biomass of the model is the following:

The Table obtained thanks to the KOA result extractor Task is the following:

We can see the different values of biomass simulated when we knockout each reaction provided in the input.
Consistency with COBRA results
If you run this code using the COBRApy module, you will reproduce the previous pipeline.
from cobra.io import load_json_model
from cobra.flux_analysis import (
single_gene_deletion, single_reaction_deletion, double_gene_deletion,
double_reaction_deletion)
ecoli_json = load_json_model('e_coli_core.json')
ecoli_json.reactions.EX_o2_e.lower_bound = 0
ecoli_json.reactions.EX_glc__D_e.lower_bound = -20
print('complete model: ', ecoli_json.optimize())
single_reaction_deletion(ecoli_json, ["ALCD2x","FUM","GLUDy", "ME2","PYK"])
The results are shown below:

We can see that the results obtained with COBRApy, a well-known and recognised module, are the same as those obtained with Constellab; our pipeline is therefore robust!
You can read this story to find out more about this Task.
